Computer-Aided Drug Discovery
In the field of computer-aided drug design, the pivotal question often revolves around determining the next optimal molecule to achieve specific property criteria. Two primary approaches guide this quest:
- Virtual Screening - involving a predefined set of candidates
- Generative Modeling - employs algorithms to propose entirely new molecular structures
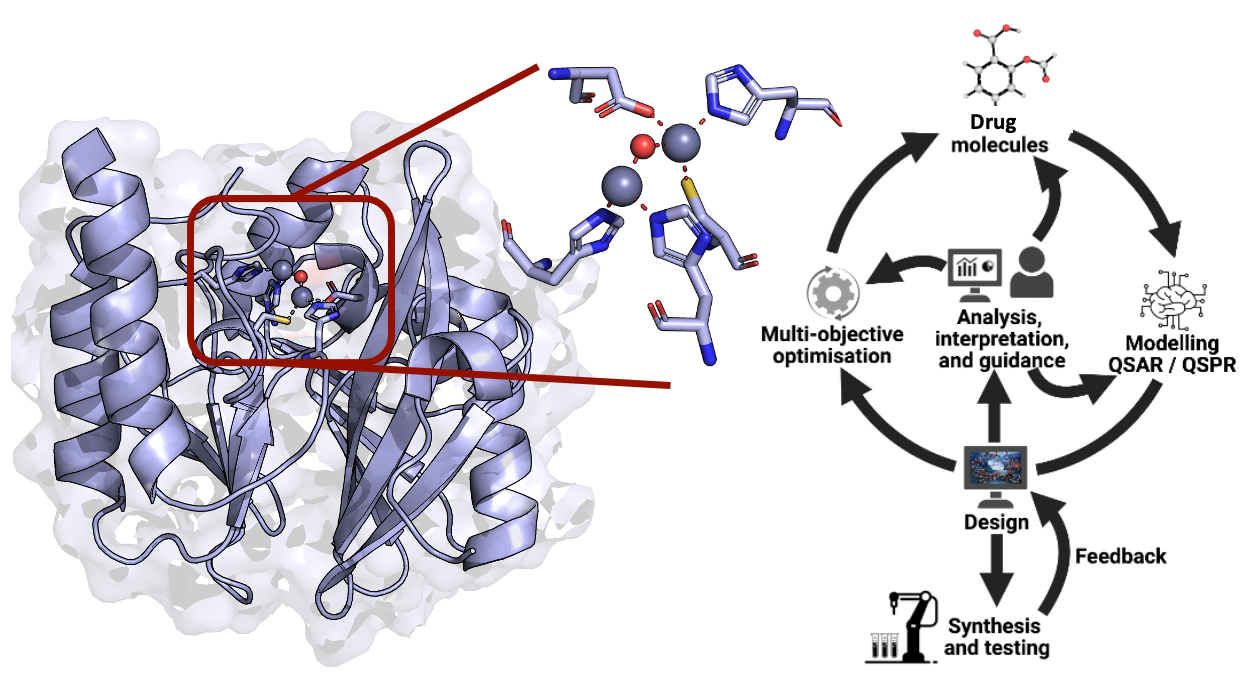
While exhaustive virtual screening, utilizing computational tools like docking for structure-based drug design, can aid in hit identification, it becomes challenging when dealing with the vast libraries encompassing billions of molecules in today's virtual landscape. Our group focuses on diverse methods for selecting and suggesting molecular structures through iterative optimization rounds. This encompasses navigating discrete design spaces of candidate molecular structures using model-guided optimisation techniques like Bayesian optimisation. A particularly intriguing thread of our research involves crafting generative models that consider synthesisability constraints, proposing molecular structures feasible for synthesis. Efficiency in sampling is also a primary concern. In the realm of in-silico benchmarks, where hundreds of thousands of molecules might be "tested," aligning with practical experimental possibilities becomes crucial, calling for models that efficiently explore these vast molecular spaces.
Related Publications →