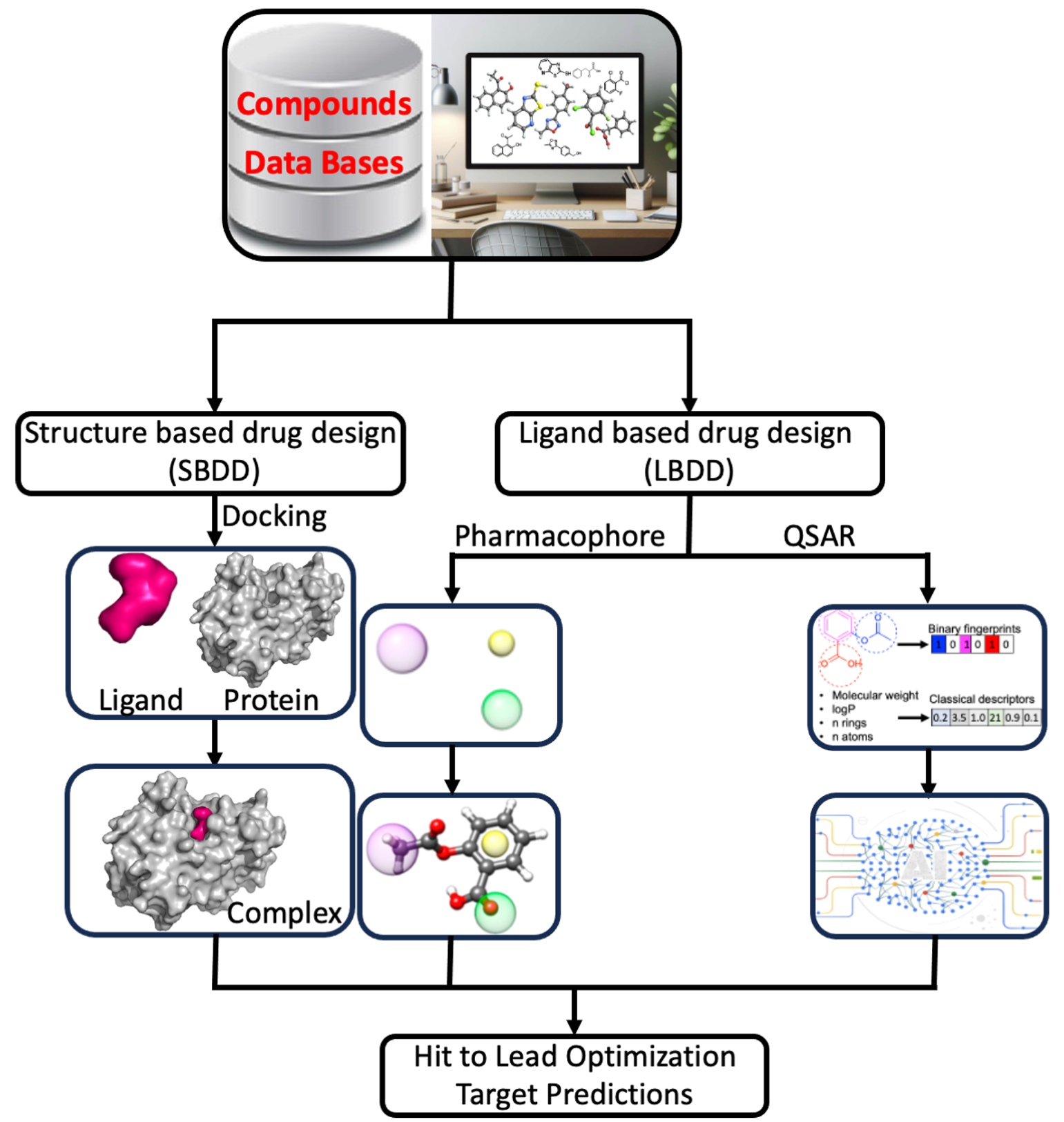
Computer-Aided Drug Design
In the realm of computer-aided drug design, the pivotal question often revolves around determining the next optimal molecule to achieve specific property criteria. Two primary approaches guide this quest: virtual screening, involving a predefined set of candidates, and generative modeling, which employs algorithms to propose entirely new molecular structures. While exhaustive virtual screening, utilizing computational tools like docking for structure-based drug design, can aid in hit identification, it becomes challenging when dealing with the vast libraries encompassing billions of molecules in today's virtual landscape. Our work focuses on diverse methods for selecting and suggesting molecular structures through iterative optimization rounds. This encompasses navigating discrete design spaces of candidate molecular structures using model-guided optimization techniques like Bayesian optimization. A particularly intriguing thread of our research involves crafting generative models that consider synthesizability constraints, proposing molecular structures feasible for synthesis. Efficiency in sampling is also a primary concern. In the realm of in silico benchmarks, where hundreds of thousands of molecules might be "tested," aligning with practical experimental possibilities becomes crucial, calling for models that efficiently explore these vast molecular spaces.
Key Publications (see all)
■ Chemical Science 2025, 16, 9691-9704.
■ European Journal of Medicinal Chemistry 2025, 291, 117584.
■ International Journal of Biological Macromolecules 2023, 253, 127379.
■ Journal of Biomolecular Structure and Dynamics 2023, 0, 1-18.
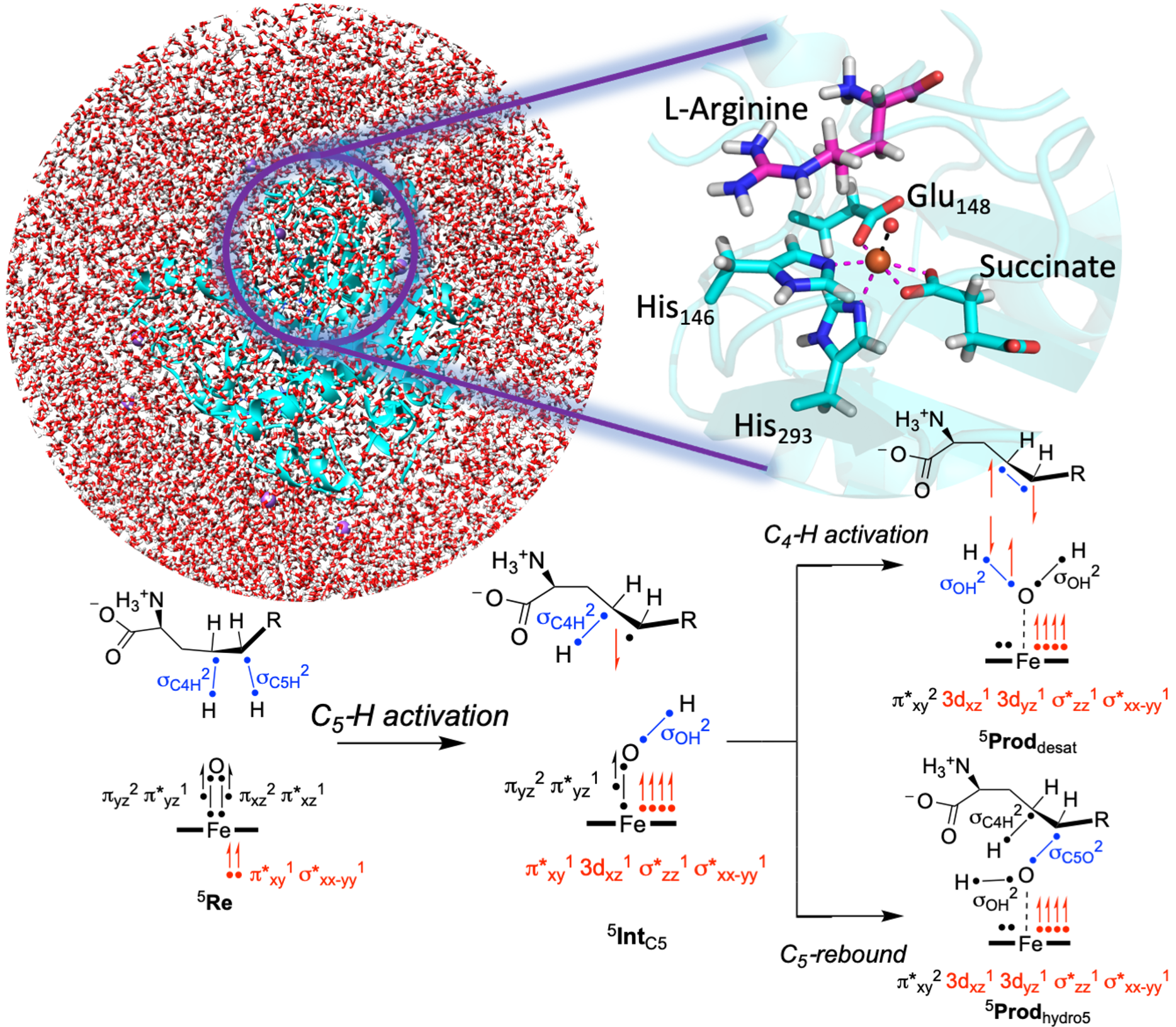
Metalloenzymes
Nature's array of metalloenzymes harnessing molecular oxygen via iron centers presents a fascinating landscape. Among these, a significant cohort harnesses a heme central ligand, while numerous others rely on non-heme ligands, showing the variety in enzymatic systems. These enzymes frequently use an oxo-iron(IV) species for oxidation reactions, extensively studied in the nonheme enzyme taurine/alpha-ketoglutarate dioxygenase. However, this species remains elusive within cytochromes P450 due to its short lifespan, making experimental studies in this field challenging. This complexity leads to a strong dependence on theoretical modeling, a key part of my research. It offers invaluable insights into pivotal queries:
> What constitutes the active oxidant species within these diverse enzymes? > How precisely do these mechanisms coordinate and activate substrates for oxidation?Through the lens of theoretical modeling, our endeavor delves deep into unraveling these fundamental mysteries, peering into the heart of enzymatic catalysis. My research journey has been marked by numerous detailed studies employing density functional theory (DFT) and quantum mechanics/molecular mechanics (QM/MM) methodologies, exploring the intricate nature of substrate oxidation catalyzed by both heme and nonheme enzymes. These investigations stand as testament to the quest for unlocking the secrets held by these remarkable biological catalysts.
Key Publications (see all)
■ ACS Catalysis 2024, 14, 11584–11590.
■ Chemistry: A European Journal 2024, 30, e202304172
■ ACS Catalysis 2023, 13, 10705–10721.
■ Journal of the American Chemical Society 2020, 142, 15764–15779.
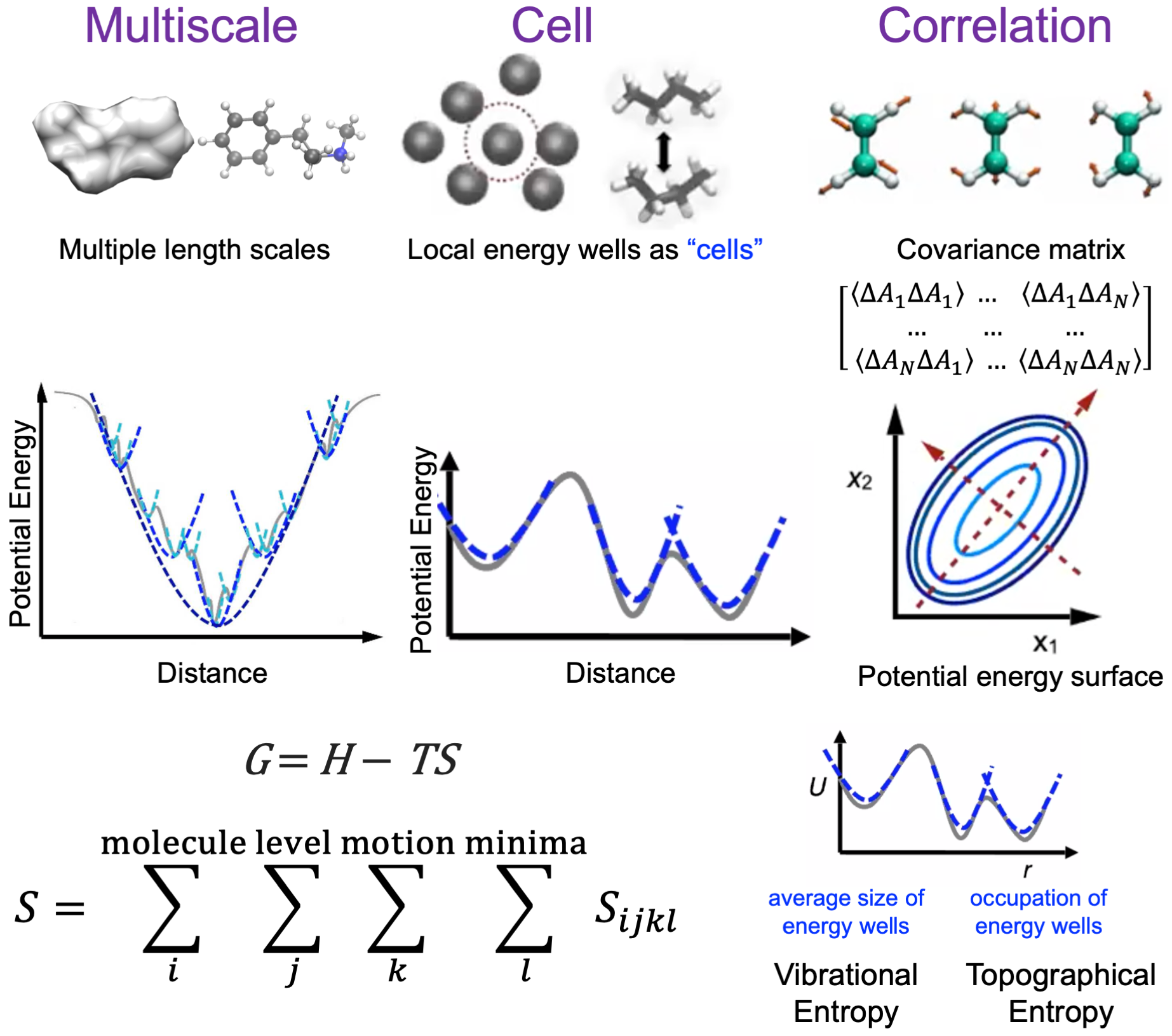
Method Development
Accurately calculating the Gibbs free energies of biomolecules in aqueous phase solution is an important challenge and future goal because most processes take place in aqueous solution. Gibbs free energies give the information about the stability and kinetics of biomolecules which will be helpful in understanding the structure and functions of biomolecules. We have developed Energy-Entropy (EE) method based on Multiscale Cell Correlation (MCC) theory. EE-MCC is a general, accurate and scalable way to quantify molecular structure and calculate its Gibbs free energy. In EE-MCC a system is divided up into cells comprising sets of particles at a hierarchy of different length scales, with correlations being accounted for particles within each cell. Probability distributions for the particles in each cell are calculated from simulation-derived forces and contacts. Of particular value is that EE-MCC makes clear the contributions of all their degrees of freedom to entropy and thus to stability.
Key Publications (see all)
■ Physical Chemistry Chemical Physics 2023, 25, 27524-27531.
■ Journal of Computer-Aided Molecular Design 2021, 35, 911–921.
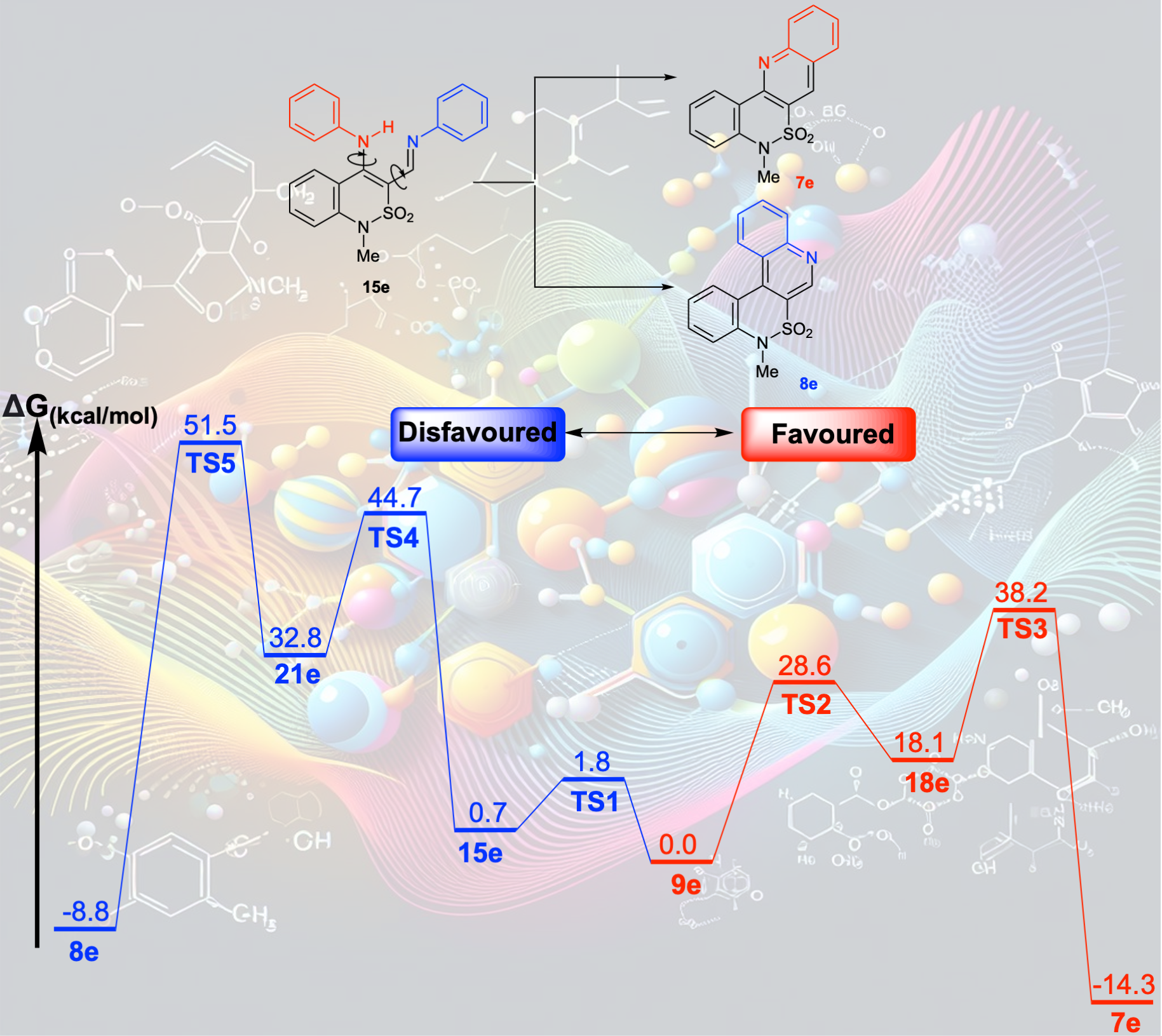
Physical Organic Chemistry
In the realm of physical organic chemistry, my approach involves harnessing Density Functional Theory (DFT) and ab initio quantum chemical calculations. These tools serve as windows into the reactivity of unconventional organic structures, unraveling the underlying reasons behind regio- and stereoselectivity in chemical reactions. This knowledge forms the basis for developing novel reactivity models, steering synthetic endeavors. These applications span diverse areas, including exploring enantioselective catalysis, and gaining a deeper comprehension of catalytic pathways. By leveraging these computational methods within the context of physical organic chemistry, we decode the intricate mechanisms guiding chemical reactions, paving the way for advancements in synthetic strategies and the understanding of complex catalytic processes.
Key Publications (see all)
■ RSC Advances 2023, 13, 2896-2902.
■ European Journal of Inorganic Chemistry 2023, 26, e202200621.